

[পূবর্ের লেকচার] [নিবন্ধন ফর্ম] [কোর্সের মূল পাতা]
আজকে আমরা পড়বো প্রোটিনের গঠনকে কিভাবে শ্রেণীবিন্যাস করা হয় এবং গঠনের অনুমান কিভাবে করা যায় এসব নিয়ে।
শ্রেণীবিন্যাস:
তিনটা ডাটাবেইজ প্রোটিনের গঠনকে বিভিন্নভাবে শ্রেণীবিন্যাস করেছে। ডাটাবেইজ তিনটি হল:
CATH (http://www.cathdb.info/)
এটা হল ম্যানুয়াল এবং অটোমেটেড দুই উপায়েই করা শ্রেণীবিন্যাসের পদ্ধতি। এর মূল চারটি ধাপ।
Class (C) – দ্বিতীয় পর্যায়ের গঠনের উপর নির্ভর করে
Architecture (A) – দ্বিতীয় পর্যায়ের গঠন কিভাবে সাজানো তার উপর নির্ভর করে
Topology (T) – দ্বিতীয় পর্যায়ের কয়টি কিভাবে নিজেদের মধ্যে যোগাযোগ করে তার উপর নির্ভর করে
Homologous superfamily (H) – গঠন এবং কাজের বিবর্তনিক সম্পর্কের উপর নির্ভর করে

CATH এর শ্রেণীবিন্যাস
SCOP (http://scop.bic.nus.edu.sg/)
এটা হল পুরোপুরি ম্যানুয়াল উপায়ে করা শ্রেণীবিন্যাসের পদ্ধতি। এর মূল তিনটি ধাপ।
Family – ৩০ শতাংশের বেশি বিন্যাসে মিল থাকা প্রোটিনগুলির বিবর্তনিক সম্পর্কের উপর নির্ভর করে
Superfamily – গঠন/কাজের সম্পর্ক থেকে ফ্যামিলি’র বিবর্তনিক উৎসের উপর নির্ভর করে
Fold – দ্বিতীয় পর্যায়ের গঠনের মূল গাঠনিক মিলগুলির উপর নির্ভর করে

SCOP অনুযায়ী শ্রেণীবিন্যাস
FSSP (http://protein.hbu.cn/fssp/)
গঠনের অনুমান:
প্রশ্ন আসতে পারে প্রোটিনের গঠনের অনুমান কেন দরকার। প্রধান কারনগুলি হল (ভিডিও লেকচারে একটু বিস্তারিত কথা বলা আছে)-

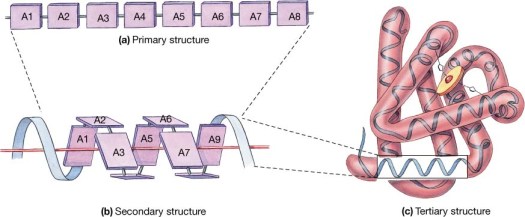
আমাদের অভিজ্ঞতা বলছে সম্ভব। কিন্তু তাতে অনেক জটিল গাণিতিক, পদার্থবৈজ্ঞানিক এবং রাসায়নিক হিসাব নিকাশের মধ্য দিয়ে যেতে হয়। কম্পিউটেশনাল বায়োলজিষ্টরা জটিল জটিল প্রোগ্রাম তৈরি করছেন শুধু নিখুঁতভাবে গঠন অনুমানের জন্য যা পরীক্ষালব্ধ গঠনের মতই হবে। কিভাবে গঠন অনুমান করা হয় তার একটা উদাহরণ এখানে দিচ্ছি।
প্রথমেই আমরা একটা এমন প্রোটিন নেই যার গঠন পরীক্ষালব্ধভাবে জানা আছে। তাহলে আমরা জানি প্রোটিনটির কোন অংশের সঙ্গে কোন অংশ সম্পর্ক তৈরি করছে। এরকম কিছু গুরুত্বপূর্ণ জায়গা (এমিনো এসিড বিন্যাস এবং পুরো বিন্যাসে অবস্থান) চিহ্নিত করি। এদেরকে বলছি মোটিফ। নিচের ছবির a তে লুপ আকারটিতে দেখুন লাল বল দুইটি। এবার প্রোটিনটির মত একইধরনের অন্য প্রোটিন খুঁজে বের করি, প্রাথমিক গঠন BLAST করলেই পাওয়া যাবে। মোটিফগুলি যদি খুঁজে পাওয়া বিন্যাসগুলিতেও সংরক্ষিত থাকে তাহলে আমরা জানছি যে এই সবগুলি প্রোটিনেই তারা একইরকম গাঠনিক বিন্যাসে থাকবে। তাহলে গঠন জানা প্রোটিনটিতে এই মোটিফগুলির অবস্থানের ভিত্তিতে বাকি প্রোটিনগুলির গঠনটা অনুমানের চেষ্ট করা যায়। আবার এমন কোন মোটিফ গঠনজানা প্রোটিনটিতে খুঁজে পাওয়া যেতে পারে যেখানে অন্যন্য অনেক মোটিফ সম্পর্ক তৈরি করে। একে বলা যায় হটস্পট। এরকম সিগনেচার হটস্পট গঠন না জানা প্রোটিনে খুঁজে পেলেও গঠন অনুমানে সুবিধা হয়। নিচের ছবি দেখুন।
তারপর এমন মোটিফগুলির সম্পর্ক থেকে বারবার প্রতিটি এমিনো এসিডের অবস্থান নিশ্চিত করে সিমুলেশানের মাধ্যমে গঠনে পৌছেন বিজ্ঞানীর। নিচের ছবি থেকে ধাপে ধাপে বিষয়গুলি দেখে নিন।

আবার শুধু এভাবে প্রোটিনের ফোল্ডই অনুমান করা যায়না। বরং অন্য প্রোটিনের সাথে গাঠনিক সম্পর্ক, গাঠনিক পরিবর্তন, লাইগেন্ড বন্ধন, কোষে কার্যক্রমের সম্পর্ক এবং পথ ইত্যাদিও অনুমান করা সম্ভব এরকম বায়োইনফরমেটিক উপায়ে। নিচের ছবিটি দেখুন।

এখন যেসব পদ্ধতি গঠন অনুমানে ব্যবহার করা হয় তার কিছু বিবরণ দিচ্ছি:
